Механизмы развития эпилепсии височной доли: клинические и экспериментальные исследования
Балыкова О.П., Шиханов Н.П., Иноземцева B.C., Сосунов А.А., МакКханн Г., Челышев Ю.А.
Колумбийский университет, г.Нью-Йорк, США
Мордовский государственный университет им. Н.П.Огарева, г.Саранск
Казанский государственный медицинский университет
Среди многих форм эпилепсии одной из наиболее изученных является эпилепсия височной доли (височная эпилепсия), связанная с патологией лимбической системы, и прежде всего гиппокампа. Источником эпилептических приступов при этой форме болезни служат отделы лимбической системы, что подтверждается электроэнцефалографическими данными, в том числе полученными с использованием внедренных электродов [81], и клинической эффективностью оперативного вмешательства. Удаление определенных отделов медиальной височной коры, в том числе части гиппокампа, способствует излечению или уменьшению частоты и тяжести приступов [92]. На основании структурных изменений выделяют два основных вида эпилепсии височной доли: 1) с наличием объемного процесса (опухоль, врожденная патология, аневризма кровеносного сосуда, кровоизлияние), затрагивающего лимбическую систему; 2) без наличия четко верифицированных объемных изменений в области медиальной височной доли [23]. В последнем случае единственным структурным проявлением височной эпилепсии является склероз гиппокампа. Название отражает наиболее яркие морфологические проявления болезни - утрату нейронов прежде всего в СА1 и СА3 зонах аммонова рога и развитие заместительного глиоза. Прижизненная визуализация мозга с использованием функциональной позитронно-эмиссионной томографии, магнито-резонансного сканирования, магнитоэнцефалографии подтверждает изменения гиппокампа при височной эпилепсии обычно в виде уменьшения его объема [60]. Наблюдается также положительная корреляция между прижизненными структурными и биохимическими (в частности число АМРА-А рецепторов и интенсивность поглощения F-флуоро-2-деокси-D-глюкозы) изменениями в склерозированном гиппокампе и данными исследования операционного материала [75].
Типичной морфологической чертой склероза гиппокампа является перестройка синаптических связей - в первую очередь изменения в ветвлении аксонов клеток-зерен зубчатой извилины с образованием обратных коллатералей, формирующих синапсы с дендритами этих же клеток во внутреннем молекулярном слое, что обозначается как спраутинг моховидных волокон (СМВ) [1]. Наличие ярких и довольно однотипных морфологических изменений в гиппокампе при эпилепсии порождает принципиальный вопрос - что первично? Являются ли изменения гиппокампа причиной развития эпилепсии и судорожных припадков или же гибель нейронов и межнейронные перестройки в гиппокампе - следствие многократно повторяющихся периодов перевозбуждения и судорог? Многочисленные клинико-морфологические и эпидемиологические исследования не позволяют однозначно ответить на этот вопрос, а их результаты легли в основу двух концепций [12, 47, 80]. Согласно одной из них, гибель нейронов и развитие глиоза происходят в результате длительно повторяющихся судорог, источником которых могут быть различные отделы головного мозга. Вторая концепция предполагает, что склероз гиппокампа является следствием патологического воздействия во время родов или в раннем детстве и в последующем приводит к развитию эпилепсии. Использование магнито-резонансного сканирования у детей с лихорадочным судорожным синдромом показало большое разнообразие изменений: наряду с отсутствием видимой патологии наблюдались признаки склероза гиппокампа, врожденная патология лимбической области, отек гиппокампа [80]. Дальнейшее наблюдение над детьми, у которых вначале определялся отек гиппокампа, а судороги продолжали регистрироваться длительное время после первоначального наблюдения, особенно при повышении температуры, показало развитие атрофии гиппокампа [80]. Сторонники обоих концепций согласны с тем, что длительный эпилептический анамнез сопровождается, как правило, более выраженными морфологическими перестройками в гиппокампе. Таким образом, подтверждается известное высказывание французского невропатолога У.Говерса (1881): «Судороги порождают судороги».
Первоначальная причина развития перевозбуждения группы нейронов и механизмы генерализации возбуждения в гиппокампе остаются неясными и составляют центральное звено в современной эпилептологии. Большое значение в выяснении механизмов развития и прогрессирования эпилепсии имеют экспериментальные подходы, среди которых наибольшее распространение получили киндлинг, каиновая и пилокарпиновая модели [4, 49, 77]. В основе этих моделей лежит перевозбуждение нейронов, приводящее к гибели клеток (каиновая и пилокарпиновая модели) или к перестройке межнейронных связей и снижению порога возбудимости нейронов и синхронизации их возбуждения. Общим патогенетическим звеном являются кальциевая перегрузка нейронов, активация ранних генов с последующей перестройкой ионных каналов, ветвлением отростков и апоптозом [29]. Схема динамики развития основных клеточных и субклеточных изменений при экспериментальной эпилепсии представлены на рис. 1.
 |
Каиновая кислота служит агонистом каинатных рецепторов, относящихся наряду с АМРА и NMDA к ионотропным глутаматным рецепторам. Клонирование генов, ответственных за синтез субъединиц каинатных рецепторов, и получение "нокаутированных" мышей позволило показать, что в клетках-зернах и пирамидных нейронах зоны САЗ аммонова рога преобладают рецепторы с большим числом субъединицы GluR6. Именно эта субъединица ответственна за эпилептогенные эффекты каиновой кислоты [53]. Повреждение нейронов при введении каиновой кислоты во многом связано с активацией c-jun N-концевой киназой 3 (JNK 3). "Нокаутированные" мыши, не экспрессирующие эту киназу, отличаются устойчивостью к нейротоксическому влиянию каиновой кислоты [90]. Взаимосвязь GluR6 и JNK 3 зависит от белка PSD-95, находящегося в постсинаптических активных зонах и выполняющего якорную функцию для киназы, активирующей JNK 3 [68].
Значение другого вида глутаматных рецепторов- «быстрых» ионотроных АМРА рецепторов - в гибели нейронов при каиновой модели эпилепсии было установлено после выяснения молекулярной структуры этих рецепторов и механизма синтеза их субъединиц. Канал представляет собой гетеротетрамер, образованный субъединицами GluRl, GluR2, GluR3 и GluR4 в различном соотношении. Обычно ионные каналы АМРА рецепторов не пропускают ионы кальция, что обеспечивается субъединицей GluR2 (GluR-B) [61]. Способность GluR2 препятствовать прохождению ионов кальция определяется только одной аминокислотой - аргинином во втором гидрофобном трансмембранном домене. Причина такого отличия GluR2 от других субъединиц рецептора связана с посттрансляционной модификацией РНК еще до ее процессинга [71]. Этот процесс осуществляется ферментом семейства аденозин деаминаз (ADAR 1), которые способны деаминировать аденозин в инозин в участке РНК, образующим двойную спираль, одна из цепей которой сформирована нитроном, причем направленные мутации в последнем приводят к нарушению редактирования РНК. Различные нейроны могут иметь каналы с разным числом GluR2, что будет отражаться в первую очередь на их способности пропускать ионы кальция в клетку. Так, пирамидные нейроны и клетки-зерна гиппокампа содержат каналы с большим числом субъединицы GluR2, в то время как в интернейронах преобладают каналы с низким содержанием этой субъединицы [48]. Гетерозиготные и гомозиготные мыши с неполноценной субъединицей GluR2, как и "нокаутированные" мыши без ADAR отличались спонтанным развитием судорожной активности и погибали в первые недели после рождения вследствие кальциевой перегрузки нейронов [35]. Снижение экспрессии GluR2 установлено в эксперименте при воздействии каиновой кислотой [27], а также в операционном материале [8].
Изменения, развивающиеся в нейронах гиппокампа - следствие их перевозбуждения и зависят от состояния сигнальных путей нейрона. Одним из первых проявлений ответной реакции нейрона на внешнее воздействие является активация системы ранних генов. Число подобных генов довольно велико и, по-видимому, будет возрастать по мере выяснения молекулярных процессов, однако сегодня можно считать, что наиболее значимы среди них, с точки зрения развития эпилепсии, гены семейств fos, jun и krox [36, 83]. Конкретные механизмы влияния продуктов ранних генов полностью не выяснены. Показано, что у "нокаутированных" мышей без с-fos киндлинг развивается хуже, чем в контрольной линии животных, а отсутствие NGFI-A (транскрипционный фактор А, представитель семейства krox, индуцируемый фактором роста нервов (NGF) не влияет на развитие судорожной активности [93]. Поскольку гибель нейронов при эпилепсии служит одним из определяющих факторов развития болезни, нельзя не отметить, что многие ранние гены и связанные с ними компоненты сигнальных путей имеют непосредственное отношение к развитию как повреждающего, так и нейропротекторного эффекта. Так, известный транскрипционный фактор CREB имеет выраженный нейропротекторный эффект [84]. Активацией CREB при фосфорилировании аминокислотного остатка в области SER 133 авторы объясняют интересное наблюдение о защитном эффекте «обогащенной» среды обитания крыс против судорожной активности, вызванной каиновой кислотой [90].
В основе спонтанной судорожной активности нейронов могут лежать следующие взаимонеисключающие причины: 1) изменения рецепторно-ионных свойств плазматической мембраны; 2) усиление притока афферентных импульсов; 3) снижение тормозного влияния интернейронов [25]. Большая часть исследований посвящена анализу причин и механизмов снижения торможения глутаматных нейронов. Это связано как с исторической последовательностью развития концептуальных представлений, так и с тем, что многие лекарственные противосудорожные препараты являются агонистами ГАМК рецепторов. В разных отделах гиппокампа механизмы торможения нейронов могут различаться. Так, при пилокарпиновой модели эпилепсии ГАМКергическое торможение усилено в клетках-зернах зубчатой извилины, но снижено в пирамидных нейронах зоны СА1 [31]. Операционный материал, взятый у больных эпилепсией, показал, что экспрессия субъединиц ГАМК-А рецепторов различалась в пирамидных нейронах СА1 и САЗ, а также клетках-зернах зубчатой извилины, что позволяет предположить существование различий в тормозном влиянии на эти нейроны [45]. Интересные данные получены в отношении торможения в зоне СА1 аммонова рога в каиновой и пилокарпиновой моделях эпилепсии - наблюдались снижение тормозного влияния в дендритах и повышение ингибиции тела пирамидных нейронов [14]. Такую особенность авторы объясняют избирательной гибелью тормозных вставочных нейронов, что приводит к снижению уровня спонтанной возбудимости клеток.
СМВ рассматривается как одна из основных причин возникновения спонтанной судорожной активности в клетках-зернах зубчатой извилины гиппокампа. При СМВ изменяются также характеристики тормозных ГАМК-А-рецепторов (возможно, и метаботропных ГАМК-В рецепторов) клеток-зерен. Плотность рецепторов значительно повышается [32], меняются также их функциональные свойства, в том числе значительно (почти в 2 раза) повышается способность ионов цинка блокировать их активность [11]. Блокада ГАМК-А рецепторов в возвратных коллатералях моховидных волокон ионами цинка показана также на операционном материале, полученном у больных с эпилепсией [73]. Последние наблюдения послужили основанием для гипотезы о выделяющемся в большом объеме из аксонов клеток-зерен цинке как причине снижения тормозного контроля клеток-зерен и развития вследствие этого судорожной активности [15]. "Нокаутированные" мыши, не содержащие один из видов трансмембранного переносчика цинка (ZnT3), оказались более лабильными к воздействию каиновой кислоты: выраженность судорог и повреждений нейронов у животных этой линии была выше, чем в исходной [13]. Следует отметить, что аксоны клеток-зерен отличаются очень высокой концентрацией ионов цинка. Именно по идентификации цинка после открытия методики гистохимического выявления тяжелых металлов по Timm впервые было показано существование СМВ при эпилепсии [28].
ГАМК-А-рецепторы являются гетерогенными полимерами и состоят из нескольких субъединиц, соотношение которых в разных рецепторах даже одной клетки варьирует [87]. Клонирование генов и анализ экспрессии разных субъединиц в модельных условиях позволили установить некоторые закономерности работы этих рецепторов и значение конкретного соотношения субъединиц для деятельности нейронов. В клетках-зернах у животных с эпилепсией снижается уровень мРНК б1- субъединицы и повышается содержание мРНК б4- субъединицы [10]. Кроме того, высокочастотная стимуляция ГАМК-А-рецепторов может приводить к пародоксальной реакции возбуждения нейронов вместо их торможения вследствие повышения выхода бикарбонатных ионов, компенсирующих эффекты входящих ионов хлора [74].
Кроме высокого содержания цинка клетки-зерна отличаются выраженной экспрессией динорфина - белка, влияющего на многие виды опиатных рецепторов и имеющего высокую аффинность к k-опиатным рецепторам [91]. Иммуногистохимическое определение динорфина, наряду с реакцией по Timm, признается достоверным критерием СМВ [62]. Динорфин способен оказывать нейропротекторный эффект вследствие уменьшения входа ионов кальция [33] и снижения выделения глутамата при возбуждении синапсов [76]. На операционном материале при эпилепсии было показано, что при выраженном СМВ динорфин не способен блокировать Са2+-каналы клеток-зерен, по-видимому, вследствие утраты ими k-рецепторов [39], что способствует перевозбуждению и синхронизации возбуждения в этой популяции нейронов.
В чем причина развития СМВ? В последнее время особое внимание уделяют активизации нейроногенеза при эпилепсии и измененным свойствам вновь образованных клеток-зерен. В норме предшественники клеток-зерен встраиваются в зубчатую извилину, а их аксоны растут в направлении САЗ и интернейронов хилуса. При эпилепсии, как полагали, именно эти вновь образовавшиеся нейроны вследствие нарушений в росте аксонов как раз и образуют обратные коллатерали. В пилокарпиновой модели эпилепсии многие нейроны, образовавшиеся во время судорожных припадков и соответствующие по электрофизиологическим параметрам клеткам-зернам, находятся в области хилуса, отличаются высоким содержанием кальбайндина D28K, повышенной возбудимостью, а их отростки образуют синаптические контакты с клетками-зернами [18].
Следует отметить, что кальбайндин D28K является кальций-связывающим белком и наряду с парвальбумином, кальретинином, белком S100 рассматривается как белок, «защищающий» клетки от кальциевых перегрузок [5]. Существование определенных взаимоотношений между экспрессией кальций-связывающих белков и уровнем кальция в нейронах было показано при анализе соотношения кальбайндин D28- и парвальбумин-иммунореактивности и числа АМРА рецепторов с «кальций-непропускающей» субъединицей GluR2. Оказалось, что экспрессия этих белков выше в клетках с минимальным содержанием GluR2 [40].
Среди многих факторов, участвующих в перестройке синаптических связей гиппокампа при эпилепсии, большое значение в последнее время придается тканевому активатору плазминогена (тАП). Наиболее ярко его роль в деятельности нейронов была впервые показана при исследованиях длительной синаптической потенциации (ДСП) в гиппокампе [65]. Оказалось, что ген тАП относится к ранним генам и служит необходимым звеном внутриклеточной сигнальной системы в реализации поздней фазы ДСП [65]. тАП является внеклеточной сериновой протеазой, наиболее известный субстрат которой плазминоген (ПГ) расщепляется тАП с образованием активного плазмина, способного влиять на многие компоненты внеклеточного матрикса. В ЦНС тАП синтезируется нейронами и клетками микроглии, а плазминоген - только нейронами [78]. Роль тАП в генезе эпилептогенных изменений может быть двоякой: во-первых, инициация процессов, приводящих к гибели нейронов, и во-вторых, участие в перестройке межнейронных связей, в частности в СМВ. Убедительное подтверждение участия тАП и ПГ в повреждении нейронов было получено на "нокаутированных" тАП (-/-) и ПГ (-/-) мышах. У тАП (-/-) и ПГ (-/-) мышей повреждение нейронов при введении каиновой кислоты было значительно менее выраженным, чем в исходной линии [78]. Нейротоксический эффект тАП определяется нарушением связи нейронов с компонентами внеклеточного матрикса, прежде всего с ламинином, деградирующим под влиянием плазмина [86], а также лизисом молекул клеточной адгезии нейронов (МКАН) [22]. Изменения внеклеточного матрикса под влиянием плазмина оказывают непосредственное влияние на ветвление и направленный рост аксонов.
Одним из компонентов внеклеточного матрикса, ответственных за этот эффект, является DSD-l-PG/фосфакан - представитель хондроитинсульфат протеогликанов, которые служат лигандами МКАН. В результате детальных исследований на тАП (-/-) и ПГ (-/-) мышах было показано, что плазмин способен лизировать DSD-l-PG/фосфакан (но не другие представители этого класса протеогликанов, например нейрокан). DSD-l-PG/фосфакан не поддерживает рост аксонов клеток-зерен зубчатой извилины, а СМВ значительно редуцирован у тАП (-/-), но не у ПГ (-/-) мышей [89]. Авторы полагают, что при развитии СМВ тАП выделяется из конуса роста аксонов, активирует плазминоген, который в свою очередь разрушает DSD-1-PG/фосфакан, делая тем самым МКАН доступными для конусов роста, что способствует росту аксонов. Следует отметить, что имеется мощная система ингибиции тАП - так называемые серпины, в частности, протеаза нексин 1 (PN-1), нейросерпин (NSP), ингибитор активатора плазминогена (PAI-1), способные образовывать стабильные комплексы с сериновыми протеазами, блокируя их ферментативную активность. В свою очередь экспрессия серпинов во многом зависит от трансформирующего фактора роста бета 1 (TGFbl) [79].
Большое значение в СМВ играют молекулы клеточной адгезии нейронов (МКАН), связанные с полисиаловыми кислотами (пСК-МКАН). Эта форма МКАН преобладает в эмбриональном периоде и имеет большое значение в развитии цито- и миелоархитектоники нервной системы, контролируя адресную миграцию клеток и направленный рост аксонов и дендритов [67]. В зрелой ЦНС пСК-МКАН обнаруживаются только в некоторых отделах головного мозга, где сохраняется деление предшественников нервных клеток, в частности в юных клетках-зернах зубчатой извилины гиппокампа [72]. Большое значение пСК-МКАН имеет в процессах, определяющих пластичность нервной системы. При снижении их содержания (введение специфического литического фермента эндонейраминидазы) или отсутствии у животных ("нокаутированные" мыши) наблюдаются нарушения памяти, обучения, процессов длительной синпатической потенциации и торможения в гиппокампе [6, 55]. Гистологически в этих случаях отмечены усиленная арборизация моховидных волокон, их дефасцикуляция, образование большого числа синапсов нетипичной локализации в области САЗ аммонова рога [16]. При височной эпилепсии на операционном материале также установлено повышение иммунореактивности на пСК-МКАН в молекулярном слое зубчатой извилины, куда врастают возвратные коллатерали клеток-зерен [51].
Одним из возможных механизмов участия пСК-МКАН в регуляции пластичности нервной ткани может являться их способность взаимодействовать с мозговым нейротрофическим фактором (BDNF) и инициировать внутриклеточные сигнальных пути этого лиганда [54]. Ростовым факторам придается большое значение как в развитии СМВ, так и в изменениях свойств других типов нейронов гиппокампа при эпилепсии [38]. Показано, что при возбуждении клеток-зерен (при многих экспериментальных моделях эпилепсии и в операционном материале) наблюдается значительное повышение экспрессии BDNF, NGF, фактора роста фибробластов (FGF) и нейротрофина-3 (NT-3) [37]. Экспрессия BDNF начинается очень быстро после возбуждения нейронов и поэтому не без основания ген BDNF относят к ранним генам [83]. У гетерозиготных мышей с резко сниженным содержанием BDNF (+/-) и NT-3 (+/-) развитие киндлинга существенно замедляется [21, 41], однако СМВ в BDNF (+/-) мышах не отличается от контрольных нормальных мышей [41]. Показано также, что BDNF при эпилепсии способствует экспрессии нейропептида Y в клетках-зернах [58], а аппликация BDNF на срезы гиппокампа от животных со СМВ приводит к возникновению спонтанных судорожных разрядов в зубчатой извилине [69].
Параллельно с экcпрессией факторов роста при эпилепсии наблюдается повышенная экспрессия тирозинкиназных рецепторов (Trk), в частности в клетках-зернах, что позволяет предположить возможность ауто- или паракринного влияния их лигандов [21]. Основное значение при эпилепсии имеют репцепторы TrkB. Подтверждением их роли в развитии судорожного синдрома могут служить эксперименты с введением в желудочки мозга своеобразных антител, так называемых "рецепторных телец", представляющих собой дивалентные гомодимеры, имеющие места связывания для лигандов, в частности для BDNF, и выполняющих таким образом роль искусственных рецепторов, связывающих лиганд. В этом случае наблюдалось выраженное угнетение киндлинга, а введение "рецепторных телец" к другим типам рецепторов Trk не влияло на его развитие [7].
Ярким проявлением компенсаторных изменений в зубчатой извилине при эпилепсии является значительное повышение экспрессии нейропептида Y (NPY) в клетках-зернах. В норме NPY определяется только в некоторых интернейронах хилуса гиппокампа и отсутствует в клетках-зернах. При СМВ NPY обнаруживается в возвратных коллатералях аксонов и его распределение аналогично локализации осадка при реакции на тяжелые металлы по Timm [46]. Полагают, что экспрессия NPY служит защитным механизмом от кальциевой перегрузки при перевозбуждении нейронов. "Нокаутированные" мыши без NPY менее устойчивы к эпилептогенным воздействиям [19], а введение NPY в гиппокамп ингибирует судорожную активность [88]. Антисудорожное действие NPY может быть связано с влиянием как на пресинаптические NPY-Y2 рецепторы, ингибирующие выделение глутамата, так и на NPY-Y1 рецепторы (в пре- и постсинаптической структуре), уменьшающие вход ионов кальция и выделение глутамата [82]. В экспрессии NPY, как полагают, большое значение имеет BDNF и его защитный эффект во многом опосредуется NPY [66].
Большое число исследований посвящено анализу экспрессии белка GAP-43, принимающему участие в росте отростков нейронов и являющемуся маркером конусов роста. В экспериментальном и операционном материале при эпилепсии значительно возрастает экспрессия мРНК и белка GAP-43 в клетках-зернах, причем максимальный уровень иммунореактивности совпадал с областями, где развивается спраутинг аксонов клеток-зерен [63]. Экспрессия GAP-43 зависит от возбуждения нейронов и уровня ионов кальция, поскольку ингибируется введением блокаторов NMDA рецепторов [50]. В гетерозиготных мышах с повышенной экспрессией GAP-43 СМВ развивается спонтанно даже без возбуждения клеток-зерен [2].
Как уже отмечалось, избирательная гибель нейронов является типичной чертой эпилепсии. Среди многих факторов, которые могут иметь значение в повреждении нервных клеток, большое внимание уделяется оксиду азота (NO) - высокоактивному межклеточному посреднику в нервной системе. Хотя данные о роли NO в реализации судорожной активности не всегда однозначны, тем не менее большинство исследований подтверждают проэпилептогенный эффект ингибиторов NO синтазы и защитный эффект при введении субстрата (l-аргинина) для продукции NO [30]. Подобные эффекты объясняют образованием при эпилепсии большего количества NO, оказывающего нейротоксический эффект [56], а также усилением кровотока и развитием окислительного стресса с образованием реактивных форм кислорода [52].
Широкое внедрение в эксперимент генетически модифицированных линий мышей и различия между линиями животных в степени повреждения нейронов, интенсивности судорожного синдрома, развитии СМВ позволяют картировать гены, ответственные за устойчивость или лабильность линии мышей. Так, было показано, что за повышенную лабильность животных к эпилептогенному эффекту препаратов ответственны определенные области 5 и 1-й хромосом [24]. В «эпилептогенных» линиях мышей (например, DBA/2, EL, ducky, lathergic, stargaze, tottering) установлены гены и характер мутаций, которые приводят к повышенной спонтанной возбудимости и судорожной активности. Многие из этих мутаций (спонтанных или индуцированных) затрагивают ионные каналы, рецепторы или компоненты основных сигнальных путей [26, 64].
Участие астроцитов в развитии и поддержании судорожной активности нейронов привлекает большое внимание исследователей [34]. Значение астроцитов в этих процессах связано прежде всего с тем, что они, обладая системами активного и пассивного трансмембранного транспорта для многих ионов и нейроактивных агентов, как и рецепторами к большинству нейротропных веществ, играют весьма большую роль в регуляции ионного гемеостаза и уровня нейромедиаторов, в первую очередь ионов калия и глутамата, в перинейрональном межклеточном пространстве, объем которого они также контролируют. Кроме того, астроциты являются источником большого числа нейроактивных веществ - цитокинов и нейротрофических факторов, влияющих на рост и арборизацию отростков нейронов.
Основное значение в регуляции поглощения ионов калия имеют блокирующиеся барием входящие выпрямляющие калиевые каналы (I Kir). При эпилепсии в области глиоза в астроцитах снижается активность этих каналов, что может отражать уменьшение буферных свойств клеток [34]. Для буферных свойств астроцитов в отношении калия большое значение имеет протяженная межклеточная связь посредством щелевых контактов, что позволяет равномерно распределять ионы в большом объеме клеток и устранять в клеточных ансамблях перепады в концентрации этого иона вследствие нейронной активности [3]. Вопрос о способности реактивных астроцитов поддерживать калиевый баланс имеет принципиальное значение, и долгое время считалось, что компенсаторные способности реактивных астроцитов снижены. Однако последние исследования показывают, что реактивные астроциты в зоне СА1 гиппокампа после развития глиоза вследствие введения каиновой кислоты способны поглощать ионы калия в значительных количествах [85]. Кроме того, реактивные астроциты связаны между собой большим числом щелевых контактов, что увеличивает их буферную способность. Следует отметить, что повышение числа щелевых контактов отмечено также на операционном материале, полученном у больных [43]. Еще одним отличием астроцитов при эпилепсии является появление клеток с потенциалзависимыми каналами [9]. Такие астроциты обнаруживаются и в нормальных условиях и обозначаются как «активные» в отличие от «пассивных», лишенных потенциалзависимых каналов. Следует отметить, что и в норме в гиппокампе астроциты не составляют однородную популяцию - в зоне СА1 преобладают «активные», а в САЗ - «пассивные» клетки [17], что свидетельствует об участии этих видов астроцитов в защите нейронов от токсических воздействий.
Значение астроцитов при эпилепсии может проявляться в синтезе трофических факторов, участвующих в росте и арборизации отростков нейронов. Так, на операционном материале, полученном у больных с эпилепсией, показано выраженное усиление экспрессии и секреции тенасцина С, компонента внеклеточного матрикса, необходимого для направленного роста и ветвления аксонов [70]. Максимальное повышение иммунореактивности зарегистрировано в области ветвления аксонов зернистых нейронов зубчатой извилины. Астроциты также могут экспрессировать пСК-МКАН [59], которые, как уже отмечалось, определяют рост аксонов клеток-зерен. Кроме того, астроциты могут синтезировать нейротрофические факторы семейства транформи-рующего фактора роста бета (TGF-beta), в частности нейртурин (NTN) и глиальный нейротрофический фактор (GDNF) [42, 44]. При развитии киндлинга экспрессия этих факторов повышается, а "нокаутированные" мыши без рецептора к этим факторам оказались более устойчивыми к развитию судорожной активности [57].
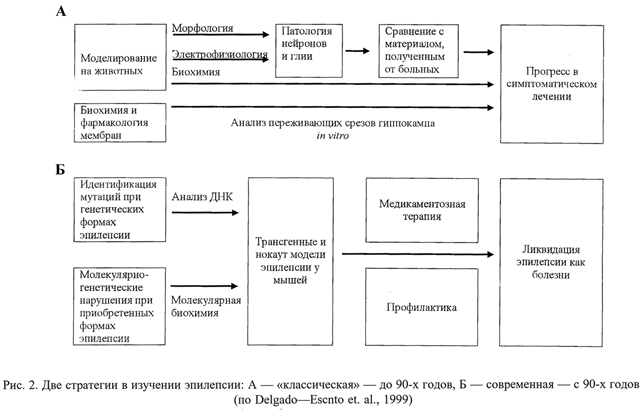
Накопленные многочисленные данные не позволяют однозначно утверждать, что то или иное изменение является необходимым условием развития спонтанной судорожной активности в гиппокампе. По-видимому, в основе возникновения эпилептических приступов лежит сочетание многих факторов и условий, причем в разных случаях (вид животного, использованная модель, клинический случай) могут быть разные сочетание и/или преобладание того или иного ведущего звена. Весомые достижения в генетике, молекулярной и клеточной биологии и межклеточных взаимоотношений принципиально меняют методологию научных исследований в области эпилепсии (рис. 2) и позволяют надеяться на прогресс не только в познании механизмов, но и в терапии эпилептического судорожного синдрома. Как одно из последних достижений в этом направлении можно выделить успешное экспериментальное использование вакцины, состоящей из аденовируса и связанного с ним гена субъединицы NR1 NMDA-рецептора [20]. Пероральное введение вакцины крысам приводило к выработке аутоантител к данной субъединице рецептора и способствовало нейропротекторному эффекту при эпилепсии и ишемии головного мозга.
 |
Литература
1. Чепурнов С.А., Чепурнова Н.Е. // Успехи физиол. наук. - 1997.- Т. 28. - С.3-50.
2. Aigner L., Arber S., Kapfhammer J.P. et al. // Cell. - 1995. - Vol. 83. - P.269-78.
3. Araque A., Parpura V., Sanzgiri R.P. et al. // Trends Neurosci. - 1999. - Vol. 22. - P.208-215.
4. Avanzini G., Moshe S.L., Schwartzkroin PA. et al. Animal models of localization-related epilepsy. In: Epilepsy: A comprehensive textbook. / Ed. J. Engel and T.A. Pedley. - Philadelphia, Lippincott-Raven Publishers, 1997. - P. 427-442.
5. Baimbridge K.G., Celio M.R., Rogers J.H. // Trends Neurosci. - 1992. - Vol. 15. - P.303-308.
6. Becker C.G., Artola A., Gerardy-Schahn R. et al. // J. Neurosci. - 1996. - P.143-152.
7. Binder D.K., Routbort M.J., McNamara J.O. // J. Neurosci. - 1999. - Vol. 19. - P.4616-4626.
8. Blumcke L, Beck H., Scheffler B. et al. // Acta Neuropathol. - 1996. - Vol. 92. - P.576-87.
9. Bordey A., Sontheimer H. // Epilepsy Res. - 1998. - Vol. 32. - P.286-303.
10. Brooks-Kayal A.R., Shumate M.D., Jin H. et al. // J. Neurosci. - 1999. - P.8312-8318.
11. Buhl E.H., Otis T.S., Mody I. // Science. - 1996. - Vol. 271. -P.369-373.
12. Cole AJ. // Epilepsia. 2000. - Vol. 41. - P.S13-22.
13. Cole T.B., Robbins C.A., Wenzel HJ. et al. // Epilepsy Res. - 2000. - Vol. 39. - P.53-69.
14. Cossart R., Dinocourt C., Hirsch J.C. et al. // Nat. Neurosci. - 2001. - Vol. 4. - P.52-62.
15. Coulter D.A. // Epilepsia. - 2000. - Vol. 41. - P.S96-99.
16. Cremer H., Chazal G., Lledo P.M. et al. // Int. J. Dev. Neurosci. - 2000. - Vol. 18. - P.213-220.
17. D'Ambrosio R., Wenzel J., Schwartzkroin P.A. et al. // J. Neurosci. - 1998. - Vol. 18. - P.4425-4438.
18. Dashtipour K., Tran P.H., Okazaki M.M., Nadler J.V., Ribak C.E. // Brain Res. - 2001. - Vol. 890. - P.261-271.
19. DePrato Primeaux S., Holmes P.V., Martin RJ. et al. // Neurosci. Lett. - 2000. - Vol. 287. - P.61-64.
20. During M.J., Symes C.W., Lawlor P.A. et al. // Science. - 2000. - Vol. 287. - P.1453-1460.
21. Elmer E., Kokaia M., Ernfors P. et al. // Exp. Neurol. - 1997. - Vol. 145. - P.93-103.
22. Endo A., Nagai N., Urano T. et al. // Neurosci Res. -1999.- Vol. 33.-P. 1-8.
23. Engel J. Jr. // Epilepsy Res. -1996. - Vol.26. - P.141-150.
24. Ferraro T.N., Golden G.T., Smith G.G. et al. // J. Neurosci. - 1999. - Vol. 19. - P. 6733-6739.
25. Fisher P.D., Sperber E.F., Moshe S.L. // Brain Dev. - 1998. - Vol. 20. - P.563-573.
26. Fletcher C.F., Frankel W.N. // Hum. Mol. Genet. - 1999. - Vol. 8.- P.1907-1912.
27. Friedman L.K., Pellegrini-Giampietro D.E., Sperber E.F. et al. // J. Neurosci. - 1994. - Vol. 14. - P.2697-2707.
28. Frotscher M., Zimmer // J Сотр. Neurol. - 1983. -Vol. 215. - P.299-311.
29. Fujikawa D.G., Shinmei S.S., Cai B. // Epilepsia. - 2000. - Vol. 41.-P.9-13.
30. Gabriel C, Friguls В., Sureda F.X. et al. // J. Neurosci. Res. - 2000. - Vol. 59. - P.797-805.
31. Gibbs J.W. 3rd, Shumate M.D., Coulter D.A. // J. Neurophysiol. - 1997. - Vol. 77. - P.1924-1938.
32. Gibbs J.W. 3rd, Sombati S., DeLorenzo R.J. et al. // J. Neurophysiol. - 1997. - Vol. 77. - P.2139-2152.
33. Hauser K.F., Foldes J.K., Turbek C.S. // Exp. Neurol. - 1999. - Vol. 160. - P.361-375.
34. Heinemann U., Gabriel S., Jauch R. et al. // Epilepsia. - 2000. - Vol. 41. Suppl 6. - P.185-189.
35. Higuchi M., Maas S., Single F.N. et al. // Nature. - 2000. - Vol. 406. - P.78-81.
36. Hughes P.E., Alexi Т., Walton M. et al. // Prog. Neurobiol. 1999. - Vol. 57. - P.421-450.
37. Inoue Т., Hirai H., Onteniente B. et al. // Neuroscience. - 1998. - Vol. 86. - P.723-728.
38. Jankowsky J.L., Patterson PH. // Prog. Neurobiol. - 2001. - Vol. 63. -P.125-149.
39. Jeub M., Lie A., Blumcke I. et al. // Neuroscience. - 1999. - Vol. 94. - P.465-71.
40. Kondo M, Okabe S, Sumino R, Okado H. // Eur. J. Neurosci. - 2000. - Vol. 12. - P.2812-2822.
41. Kokaia M., Ernfors P., Kokaia Z. et al. // Exp. Neurol. - 1995. - Vol. 133. - P.215-24.
42. Kotzbauer P.T., Lampe P.A., Heuckeroth R.O. et al. // Nature. - 1996. - Vol. 384. - P.467-470.
43. Lee S.H., Magge S., Spencer D.D. et al. //. Glia. - 1995. - Vol. 15. - P.195-202.
44. Lin L.F., Doherty D.H., Lile J.D. et al. //. Science. - 1993.-Vol. 260. - P.1130-1132.
45. Loup F, Wieser H.G., Yonekawa Y. et al. // J. Neurosci. - 2000. - Vol. 20. - P.5401-5419.
46. Makiura Y, Suzuki F, Chevalier E., Onteniente B. // Exp. Neurol. - 1999. - Vol. 159. - P.73-83.
47. Mathern G.W, Babb Т.Е., Armstrong D.L. Hippocampal sclerosis. In: Epilepsy: A comprehensive textbook. / Eds. J. Engel and T.A. Pedley. - Philadelphia, Lippincott-Raven Publishers, 1997.-P.133-155.
48. McBain C.J., Fisahn A. // J. Neurosci. - 1994. - Vol. 14. - P.3413-3425.
49. McNamara, J.O. , Bonhaus, D.W., Shin C. Epilepsy: Models, Mechanisms and Concepts. / Ed. P.A. Schwartzkroin. -Cambridge Univ. Press, Cambridge, U.K., 1993. - P.27-47.
50. McNamara R.K., Routtenberg A. // Mol. Brain Res. - 1995. - Vol. 33. - P.22-28.
51. Mikkonen M., Soininen H., Kalvianen R. et al. // Ann. Neurol. 1998. - Vol. 44. - P.923-934.
52. Montecot C, Rondi-Reig L., Springhetti V et al. // Neuroscience. - 1998. - Vol. 84. - P.791-800.
53. Mulle C., Sailer A., Perez-Otano I. et al. // Nature. - 1998. - Vol. 392. - P.601-605.
54. Muller D., Djebbara-Hannas Z., Jourdain P. et al. // Proc. Natl. Acad. Sci. USA. - 2000. - Vol. 97. - P.4315-4320.
55. Muller D., Wang C, Skibo G. et al. // Neuron. - 1996. - Vol. 17.- P.413-422.
56. Mulsch A., Busse R., Mordvintcev P.I. et al. // Neuroreport. - 1994. - Vol. 5. - P.2325-2328.
57. Nanobashvili A., Airaksinen M.S., Kokaia M. et al. // Proc. Natl. Acad. Sci. USA. - 2000. - Vol. 97. - P.12312-12317.
58. Nawa H., Pelleymounter M.A., Carnahan J. // J. Neurosci. - 1994. - Vol.14. - P.3751-3765.
59. Nomura Т., Yabe Т., Rosenthal E.S. et al. // J. Neurosci. Res. - 2000. - Vol. 61. - P.588-596.
60. Otsuki Т., Yoshimoto T. // Epilepsia. - 2000. - Vol. 41. Suppl 9. - P.26-27.
61. Pellegrini-Giampietro D.E., Gorter J.A., Bennett M.V., Zukin R.S. // Trends Neurosci. - 1997. - Vol. 20. - P.464-470.
62. Pierce J.P., Kurucz O.S., Milner T.A. // Hippocampus. - 1999. - Vol. 9. - P.255-276.
63. Proper E.A., Oestreicher A.B., Jansen G.H. et al. // Brain. - 2000 - Vol. 123. - P.19-30.
64. Puranam R.S., McNamara J.O. // Curr. Opin. Neurobiol. - 1999. - Vol. 9. - P.281-287.
65. Qian Z., Gilbert M.E., Colicos M.A. et al. // Nature. - 1993. - Vol. 361. - P. 453-457.
66. Reibel S., Larmet Y, Carnahan J. et al. // Epilepsia. - 2000. - Vol. 41. Suppl 6. - P. 127-133.
67. Rutishauser U., Landmesser L. // Trends Neurosci. - 1996. - Vol. 19. - P. 422-427.
68. Savinainen A., Garcia E.P., Dorow D et al. // J. Biol. Chem. - 2001 (№M 100190200 в электронной версии)
69. Scharfman H.E., Goodman J.H., Sollas A.L. // J. Neurosci. - 1999.- Vol. 19. - P.5619-5631.
70. Scheffler В., Faissner A., Beck Het al. // Glia. - 1997. - Vol. 19. - P.35-46.
71. Seeburg PH., Higuchi M., Sprengel R. // Brain Res. Rev. - 1998. - Vol. 26. - P.217-229.
72. Seki Т., Arai Y. // Neuroreport. - 1995. - Vol. 6. - P.2479-2482.
73. Shumate M.D., Lin D.D., Gibbs J.W. 3rd et al. // Epilepsy Res.- 1998.-Vol. 32.- P.114-128.
74. Staley K.J., Soldo B.L, Proctor W.R. // Science. - 1995. - Vol. 269.- P.977-981.
75. Szelies В., Weber-Luxenburger G., Mielke R. et al. //. Eur. J. Neurol. - 2000. - Vol. 7. - P.393-400.
76. Terman G.W., Drake C.T., Simmons M.L. et al. // J. Neurosci. - 2000. - Vol. 20. - P.4379-4388.
77. Treiman D.M., Heinemann U. Experimental models of status epilepticus. In: Epilepsy: A comprehensive textbook. / Eds. J. Engel and T.A. Pedley. - Philadelphia, Lippincott-Raven Publishers, 1997. - P.443-455.
78. Tsirka S.E., Rogove A.D., Bugge Т.Н. et al. // J. Neurosci. - 1997. - Vol. 17. - P.543-552.
79. Turgeon V.L., Houenou L.J. // Brain Res. Rev. - 1997. - Vol. 25. - P.85-95.
80. VanLandingham K.E., Heinz E.R., Cavazos J.E., Lewis D.V. // Ann. Neurol. - 1998. - Vol. 43. - P.413-426.
81. Van Roost D., Solymosi L., Schramm J. et al. // Neurosurgery. - 1998.Vol. 43. - P.819-826.
82. Vezzani A., Sperk G., Colmers W.F. // Trends Neurosci. - 1999. - Vol. 22. - P.25-30.
83. Walton M., Henderson C, Mason-Parker S. et al. // J. Neurosci. Res. - 1999. - Vol. 58. - P.96-106.
84. Walton M., Woodgate A.M., Muravlev A et al. // J. Neurochem. - 1999. - Vol. 73. - P.1836-1842.
85. Walz W, Wuttke W.A. // J. Neurosci. Res. - 1999. - Vol. 56. - P.595-603.
86. Werb Z. // Cell. - 1997. - Vol. 91. - P.439-442.
87. Whiting P.J., Bonnert T.P., McKernan R.M. et al. // Ann. NY Acad. Sci. - 1999. - Vol. 868. - P.645-653.
88. Woldbye D.P., Madsen T.M., Larsen P.J. et al. // Brain Res. - 1996. - Vol. 737. - P.162-168.
89. Wu Y.P., Siao C.J., Lu W. et al. // J. Cell Biol. - 2000. - Vol. 148. - P.1295-1304.
90. Young D., Lawlor P.A., Leone P. et al. // Nat. Med. -1999. - Vol. 5. - P.448-453.
91. Zang N., Houser C.R. // J. Сотр. Neurol. - 1999 - Vol. 405. - P.472-490.
92. Zentner J., Hufnagel A., Wolf H.K. et al. //. J. Neurol. Neurosurg. Psychiatry. - 1995. - Vol. 58. - P.666-763.
93. Zheng D., Butler L.S., McNamara J.O. // Neuroscience. - 1998. - Vol. 83. - P.251-258.
Балыкова О.П., Шиханов Н.П., Иноземцева B.C., Сосунов А.А., МакКханн Г., Челышев Ю.А. Механизмы развития эпилепсии височной доли: клинические и экспериментальные исследования // Неврологический вестник. - 2002. - Т. XXXIV, вып. 1-2. - С.60-68.